The overarching research focus of our group centers on leveraging materials informatics and data-driven methodologies to advance high-impact research in the realms of materials design and discovery. Our endeavors encompass not only the pursuit of fundamental scientific insights but also the provision of innovative solutions to address significant challenges spanning various fields.
Our current interests are directed towards several domains, including achieving carbon neutrality, enhancing fuel production, harnessing solar energy, advancing water treatment techniques, and developing cutting-edge energy storage solutions.
Our strategic approach involves the establishment of a robust infrastructure, which encompasses the development of automated virtual high-throughput screening tools, the construction of comprehensive databases, and the implementation of AI-driven workflows tailored to address specific research inquiries.
Recent work

We developed a comprehensive computational database for C20–C60 fullerenes, covering 5770 structures and calculating 12 properties using DFT, including stability, HOMO-LUMO gap, and solubility. Our study shows that electronic properties can be tailored without affecting stability. Novel topological features predict stability more accurately than traditional rules, guiding fullerene optimization for organic solar cells.
https://www.nature.com/articles/s41524-024-01410-7
Fine the Behind the Paper Story here:
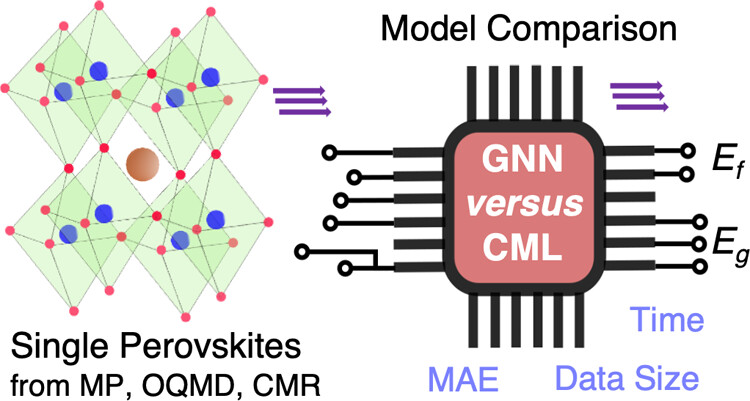
Our study evaluates machine learning (ML) models for predicting key properties—formation energy and band gaps—of perovskite materials. We benchmarked conventional ML models (CML) against graph neural networks (GNN), identifying LGBM and GATGNN as top performers for their balance of accuracy and efficiency. Using over 1000 data points, we achieved optimal prediction results and leveraged SHAP analysis for better interpretability of CML models. Our work establishes a standardized benchmark, guiding future model selection and advancing the discovery of perovskite materials in energy and electronic applications.
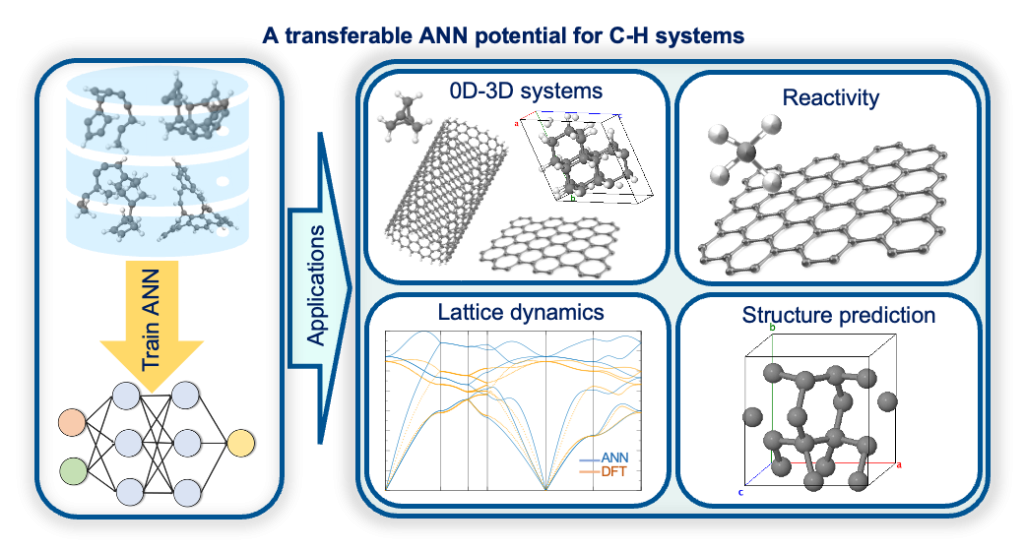
We developed a cutting-edge machine learning interatomic potential (MLIP) using artificial neural networks (ANN) to model carbon–hydrogen (C–H) systems. Trained on data from density functional theory (DFT) calculations, our ANN-based model excels in predicting atomic structures, energies, and lattice dynamics with remarkable accuracy and transferability. What makes this work truly exciting is its success in discovering a novel carbon polymorph, showcasing the potential of AI-driven approaches to uncover new materials. This breakthrough not only advances our understanding of C–H systems but also opens up new possibilities in materials science, demonstrating how AI can drive the discovery of advanced materials.
https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp02300e/unauth
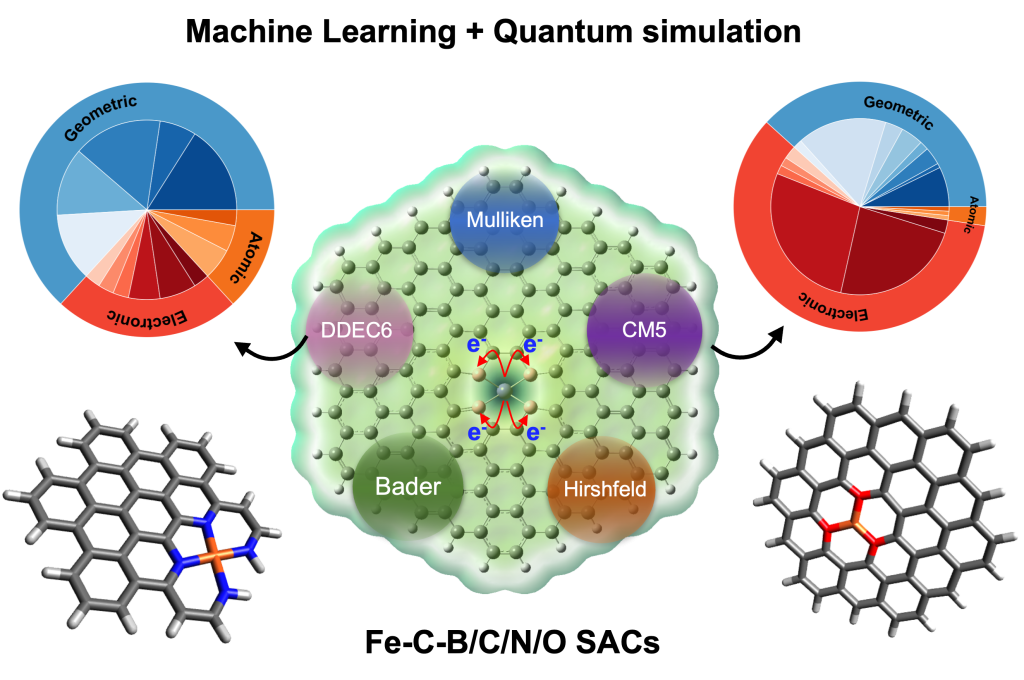
Can diverse charge schemes yield unified insights? Our study on Fe-centered SACs dives into Bader, Mulliken, Hirshfeld, CM5, and DDEC6 charges. Excitingly, DDEC6, Mulliken, and Hirshfeld show stronger correlations, impacting Fe’s atomic charge. Metal-substrate interactions sway these charges uniquely. Leveraging Quantum Simulations and Machine Learning, we predict charge schemes, revealing geometric and electronic influences. Discovering SACs’ design secrets, one charge at a time.